
Bioequivalence Between Two Tablets of Lacosamide in Healthy Subjects Under Fasting Condition

Celso Francisco Pimentel Vespasiano¹, Valery Aleksandravicius de Carvalho Accennato¹, Fernando Costa1, Leonardo de Souza Teixeira ², Fernanda Cruvinel de Abreu², Iram Moreira Mundim², Carina Pimentel Itapema Alves²
¹Zodiac Produtos Farmacêuticos S/A.– São Paulo – Brazil.
²ICF–Instituto de Ciências Farmacêuticas de Estudos e Pesquisas. Goiânia – Goiás – Brazil.
Corresponding Author: Celso Vespasiano
celso.vespasiano@zodiac.com.br
ABSTRACT
Bioavailability from different formulations of lacosamide200 mg film-coated tablets was compared in a bioequivalence study under fasting conditions. The study was single dose, randomized, open label, two-period crossover, with Brazilian males and females healthy subjects. Blood samples were taken during 72 h and plasmatic concentrations were determined using a validated HPLC-MS/MS method. Confidence intervals (CI90%) for the peak plasma concentration (Cmax) and area under the concentration-time curve (AUC0-t) were determined by calculating log-transformed data. The ratios and 90% CI for the geometric mean test/reference ratios were 96.17% (89.30-103.56%) for Cmax and 98.69% (96.71-100.72%) for AUC0-t. Under fasting condition, the test (lacosamide 200 mg film-coated tablets, Monte Verde S.A.) and reference (Vimpat® 200 mg film-coated tablets, Aesica Pharmaceuticals GmbH) formulations were considered bioequivalent since the 90% CIs for the geometric mean test/reference ratios were within ANVISA’s predetermined range of 80% to 125%.
Keywords: lacosamide, film-coated tablets, fasting conditions, bioequivalence, chromatography
1. INTRODUCTION
Lacosamide, chemical name (R)-2-acetamido-N-benzyl-3-methoxypropionamide, is a functionalized amino acid. Lacosamide has amphiphilic properties, on one hand being sufficiently water soluble to support complete and fast absorption after an oral dose, and on the other hand being sufficiently lipophilic to cross the blood–brain barrier. Lacosamide has a molecular weight of 250.3 and is classified under the Biopharmaceutical Classification System as a Class 1 compound (i.e. high solubility and high permeability) [1].
Lacosamide is indicated as monotherapy or adjunctive therapy for patients with partial-onset seizures and focal epilepsies [2, 3]. It was approved by both the FDA and the EMEA in 2008. It enhances the inactivation of voltage-gated sodium channels, leading to the stabilization of hyper-excitable neuronal membranes. Lacosamide has been proposed for the treatment of neuropathic pain of various etiologies in patients who do not respond to standard treatments [3].
Following oral administration, plasma concentrations of lacosamide increased rapidly and reached a maximum at about 0.5–4 h post dose (tmax). The absorption is complete with almost 100% bioavailability [1, 2, 3, 4]. Intake of food does not affect the area under the plasma concentration–time curve (AUC) or Cmax of lacosamide [1, 3]. A dose-proportional increase of Cmax and AUC was observed after a single oral lacosamide dose ranging from 100 to 800 mg [1, 2].
Plasma protein binding for lacosamide is low (approximately< 15 %) in healthy subjects as well as in patients with partial-onset seizures [1]. It is primarily metabolized via CYP2C19 (demethylation) to an inactive O-desmethyl metabolite (30%). Unchanged lacosamide predominates in human plasma after administration, with circulating metabolites having no known pharmacological activity [1]. Approximately 40% of lacosamide is eliminated unchanged via renal excretion mechanisms [3].
Elimination of lacosamide from plasma occurs with a terminal half-life (t½) of 12–14 h in young subjects and 14–16 h in elderly subjects [1, 2, 4]. PK analysis indicated low interindividual and intraindividual variability of PK parameters [4].
The purpose of the study in this paper was to compare, in healthy volunteers from both genders, the pharmacokinetic profiles of lacosamide, aiming to assess the bioequivalence between two formulations: lacosamide200 mg film-coated tablet, registered by Zodiac Produtos Farmacêuticos S.A. (test drug) and Vimpat® 200 mg film-coated tablet, imported by Meizler UCB Biopharma S.A. (reference drug) under fasting condition.
2. METHODS
Population
Thirty-three (33) volunteers of both genders (17 female 16 male subjects) aged between 18 and 50 years were screened for the present study. There were no restrictions for ethnic group. All volunteers were considered as being eligible to participate in the studies and fulfilled all the inclusion and exclusion criteria defined in the protocol.
All volunteers showed good health conditions or the absence of significant diseases after assessment of medical history, verification of vital signs, physical examination, electrocardiogram, and routine laboratory tests. All subjects enrolled in the studies showed negative tests for hepatitis B (HBsAg and Anti-HBc IgM), hepatitis C, HIV and urine HCG (pregnancy test only for female subjects).
The study was conducted in compliance with guidelines and standards for researches involving human beings from Resolutions no. 466/12 and 251/97 by the National Health Council – Ministry of Health, Good Clinical Practices according to ICH, and the Document of the Americas and in compliance with the Declaration of Helsinki (adopted by the 18th WMA General Assembly in Helsinki/ Finland, 1964, and with the last amendment by the 64th WMA General Assembly in Fortaleza/ Brazil, 2013). The protocol was submitted and approved before study initiation by the Research Ethics Committee of Instituto de Ciências Farmacêuticas de Estudos e Pesquisas S/S in Aparecida de Goiânia, Goiás, Brazil. After explaining the nature and purpose of the study, all volunteers provided their written informed consent for participation.
Study Treatments
In this study, the test formulation was lacosamide 200 mg film-coated tablets (batch number 76128), manufactured by Monte Verde S.A., Argentina, and the reference formulation was Vimpat®, lacosamide200 mg film-coated tablets (batch number 9333301), manufactured by Aesica Pharmaceuticals GmbH, Germany.
Study Design
The study was conducted using an open-label, randomized, two-period, crossover, and prospective design, with a washout period of 7days between administrations. In each of the study periods, the volunteers received a film-coated tablet containing 200 mg of lacosamide from one of the two formulations mentioned above orally , as a single dose with a 200-mL of water at room temperature. The drugs were administered after a minimum fast of 8 hours. Volunteers fasted for 4 hours after drug administration. In order to maintain the standardization of treatment groups, the diet (food and drink) followed the same standard for all volunteers and in both periods.
The intake of alcoholic beverages, food or beverages containing caffeine or xanthine (such as coffee, tea, chocolate and cola- or guarana-based soft drinks) was not permitted. In addition, the use of nicotine was prohibited from 24hours before hospitalizations until the last blood draw.
Blood samples (7.5 mL) were collected in coated tubes, containing lithium heparin as anticoagulant.
Study blood schedule included collections before (pre-dose) and 00:10, 00:20, 00:30, 00:45; 1:00; 1:20; 1:40; 2:00; 2:30; 3:00; 3:30; 4:00; 5:00; 6:00; 9:00; 12:00; 24:00; 36:00; 48:00, and 72:00 hours after dose administration. A total of 21 blood samples were collected from each volunteer in each period.
Right after collection, blood samples were centrifuged at 3,000 rpm for 5minutes at approximately 4°C. Immediately after centrifugation, the plasma was separated and transferred to two previously labeled tubes. The tubes were stored in a freezer at -20°C and were maintained at this temperature until the analysis.
Clinical, analytical, and statistical stages of the study were conducted by Instituto de Ciências Farmacêuticas de Estudos e Pesquisas (ICF), located in Goiás, Brazil.
Quantification of lacosamide in human plasma
Plasma concentrations of lacosamide were determined using high-performance liquid chromatography with sequential mass spectrometry (HPLC-MS/MS). The analytes were extracted from plasma using precipitation. Carbamazepine was used as the internal standard. In order to avoid inter-assay variations, all the samples from the same volunteer were assessed in the same analytical run.
The detection parameter used was the mass-to-charge ratio (m/z) between precursor ions and product, and the quantification parameter was the ratio of areas under chromatogram peak identified in the retention time between analyte and internal standard. Lacosamide concentrations in volunteer samples were calculated using interpolation in the calibration curve.
The chromatographic analysis was conducted in a HPLC Agilent 1200 Series and mass detector API 3200 MS/MS with column Inertsil ODS-4 5μm 4.6x100mm, with a flow rate of 1 mL/min. The column was maintained at 20°C. The mobile phase used was acetonitrile and 2mMammonium acetate with formic acid 0.025% at a 75:25ratio (v/v). The injection volume was 5 μL and the total run time set as 2minutes. The mass spectrometry detection was conducted using electrospray ionization source in positive mode. The multiple reaction monitoring (MRM) method was used, and the transitions monitored were m/z 251.049 > 74.2and m/z 237.12> 194.2for lacosamide and carbamazepine, respectively.
The method was validated in compliance with ANVISA guidance for bioanalytical method validation, RDC Resolution no. 27/ 2012 (5). The linearity range was from 100 to 10000 ng/mL. The validation parameters assessed were selectivity, linearity, intra- and inter-run precision, intra- and inter-run accuracy, matrix effect, residual effect, and stability of lacosamide under different conditions.
Pharmacokinetic and Statistical Analysis
The pharmacokinetic parameters were obtained from the curves of plasma concentration vs. time for lacosamide and statistically assessed for determination of bioequivalence, using Phoenix WinNonLinand Microsoft Excel softwares. The area under the curve of plasma concentration vs. time was calculated using the trapezoidal method, from time zero to the last measurable concentration (AUC0-t). The area under the curve of plasma concentration vs. time was also calculated from time zero to infinity (AUC0-∞), where AUC0-∞ =AUC0-t + Ct/Kel, with Ct being the last drug concentration experimentally defined and Kel being the terminal phase elimination rate constant. The peak of maximum plasma concentration (Cmax) of lacosamide and the time to reach this peak (tmax) were obtained directly with no data interpolation. The elimination half-life (t½) was defined using the equation t½ = ln (2) /Kel.
For the assessment of bioequivalence between the two formulations, AUC and Cmax parameters were used. A 90% Confidence Interval (CI) was generated for the difference in averages of transformed data from test and reference drugs. The antilog of obtained CI comprised the 90% CI for geometric mean ratio of primary parameters. The drugs are considered bioequivalent if the limits of CI generated for the geometric mean ratio for both primary parameters are higher than 80% and lower than 125%, as determined by ANVISA and FDA [6,7].
3. results
In the study, 30volunteers completed the two study periods out of 32 participants. The volunteers had mean age of 34 years, ranging from 23 to 49 years; mean weight of 69.33 kg (52.45 to 91 kg); mean height of 1.64 m (1.43 to 1.82) and mean BMI of 25.48 kg/m² (19.59 to 28.53) kg/m².
Lacosamide was well tolerated at the administered dose. No serious adverse events were seen or reported, and no pregnancies were detected during the study. The most common adverse event was laboratory exams abnormal results on the post-study evaluation, noticed by 53.3% of the volunteers.
The mean curve for plasma concentration vs. time obtained for test and reference drugs are shown in Figure 1. The curves were shown to be overlapped, demonstrating a similar pharmacokinetic profile between the drugs (A: reference treatment; B: test treatment).
The central location and dispersion measures for all pharmacokinetic parameters from both formulations are shown in Table 1.
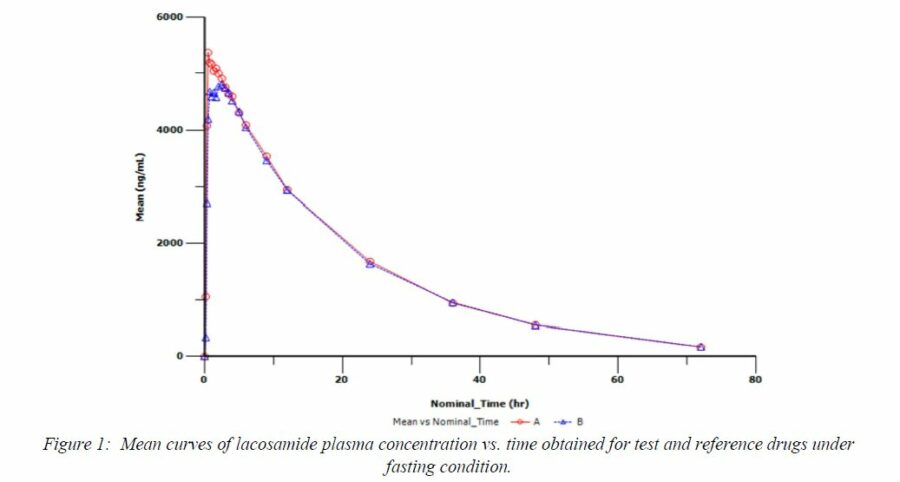
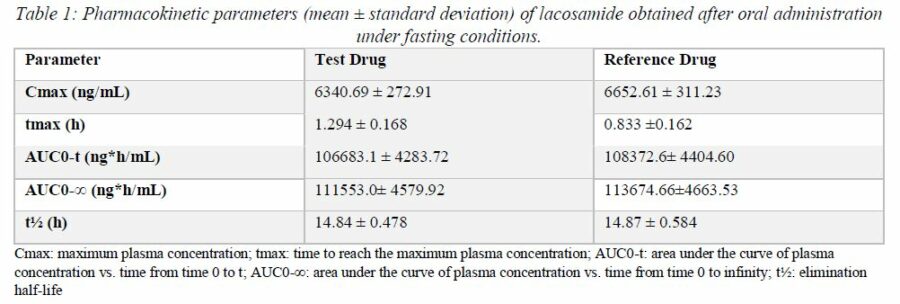
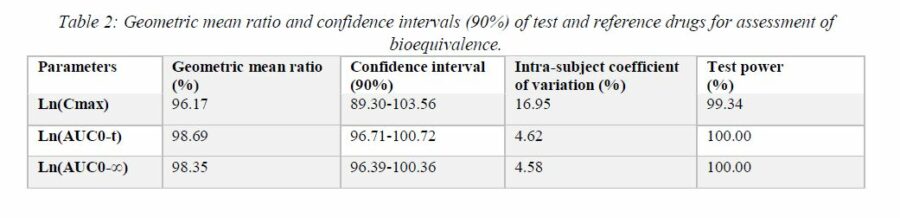
Table 2 shows the test/reference geometric mean ratios for pharmacokinetic parameters Cmax, AUC0-t, and AUC0-∞and the respective 90% CIs for the bioequivalence analysis of the study. All 90% CIs were within the interval of 80% to 125%.
4. Discussion
With the aim of obtaining a highly sensitive and rapid method for quantification of lacosamide in plasma, a method by HPLC-MS/MS was developed and validated in this project. In the presented method, the limit of quantification was 100 ng/mL, which allowed for a sensitive and efficient analysis of lacosamide plasma concentrations. Liquid chromatography-mass spectrometry (MS) has become increasingly common because of its high selectivity and sensitivity [8].
Two drugs are considered bioequivalent if their rate and extent of absorption do not show statistically significant differences when administered at the same molar dose of the active ingredient, under the same experimental conditions [9]. In this study, the relative bioavailability of two formulations of lacosamide was assessed after administration under fasting conditions.
The pharmacokinetic results (Cmax, AUC, tmax and t½) found in this study for lacosamide (Table 1) were very similar to those reported in the literature [1, 2, 3, 4]. As shown in Table 2, 90% CIs obtained for pharmacokinetic parameters defining bioequivalence (Cmax, AUC0-t, and AUC0-∞) of both formulations of lacosamide 200 mg were shown to be within the bioequivalence limits defined by ANVISA (80%-125%) in RE Resolution no. 1170/ 2006 [6].
5. conclusion
In this bioequivalence study conducted in health volunteers under fasting conditions, based on pharmacokinetic and statistical results obtained, we conclude that the test drug (lacosamide 200 mg –Monte Verde S.A.) and the reference drug (Vimpat® 200 mg – Aesica Pharmaceuticals GmbH) are bioequivalent. Thus, lacosamide film-coated tablets may be considered as being interchangeable in medical practice, since they have the same efficacy and safety profile for the patients.
REFERENCES
1. Cawello W. Clinical Pharmacokinetic and Pharmacodynamic Profile of Lacosamide. Clinical Pharmacokinetic. 2015; 54(9):901-14.
2. Cawello W, Rosenkranz B, Schmid B, Wierich W.Pharmacodynamic and pharmacokinetic evaluation of coadministration of lacosamide and an oral contraceptive (levonorgestrel plus ethinylestradiol) in healthy female volunteers. Epilepsia. 2013; 54(3):530–536.
3. Jacob S, Nair AB. An Updated Overview on Therapeutic Drug Monitoring of Recent Antiepileptic Drugs. Drugs R D. 2016; 16:303–316.
4. Cawello W, Bokens H, Nickel B, Andreas J-O, Halabi A. Tolerability, pharmacokinetics, and bioequivalence of the tablet and syrup formulations of lacosamide in plasma, saliva, and urine: Saliva as a surrogate of pharmacokinetics in the central compartment. Epilepsia. 2013; 54(1):81–88.
5. Agência Nacional de Vigilância Sanitária (Brasil). Resolução n° 27, de 17 de maio de 2012. Dispõe sobre os requisitos mínimos para a validação de métodos bioanalíticos empregados em estudos com fins de registro e pós-registro de medicamentos. Diário Oficial da União 22 mai 2012.
6. Agência Nacional de Vigilância Sanitária (Brasil). Resolução n° 1170, de 19 de abril de 2006. Guia para provas de biodisponibilidade relativa / bioequivalência de medicamentos. Diário Oficial da União 24 abr 2006.
7. United States Food and Drug Administration. Guidance for Industry: Bioavailability and Bioequivalence Studies for Orally Administered Drug Products – General Considerations. Rockville, 2003.
8. Gu J, Wu W, Huang M, Long F, Liu X, Zhu Y. Application of High-Performance Liquid Chromatography Coupled with Linear Ion Trap Quadrupole Orbitrap Mass Spectrometry for Qualitative and Quantitative Assessment of Shejin-Liyan Granule Supplements. Molecules. 2018; 23, 884.
9. Agência Nacional de Vigilância Sanitária (Brasil). Manual de Boas Práticas em Biodisponibilidade e Bioequivalência, Volume 1. Gerência-Geral de Inspeção e Controle de Medicamentos e Produtos. Brasília: ANVISA, 2002.
Sorry, the comment form is closed at this time.