
Patogenia, fatores de risco e fisiopatologia da FPI (Fibrose pulmonar idiopática)
Interação entre predisposição genética individual para o desenvolvimento de fibrose, e mecanismos relacionados ao envelhecimento e fatores ambientais constitui a teoria hoje mais aceita para o desenvolvimento da FPI.9, 10
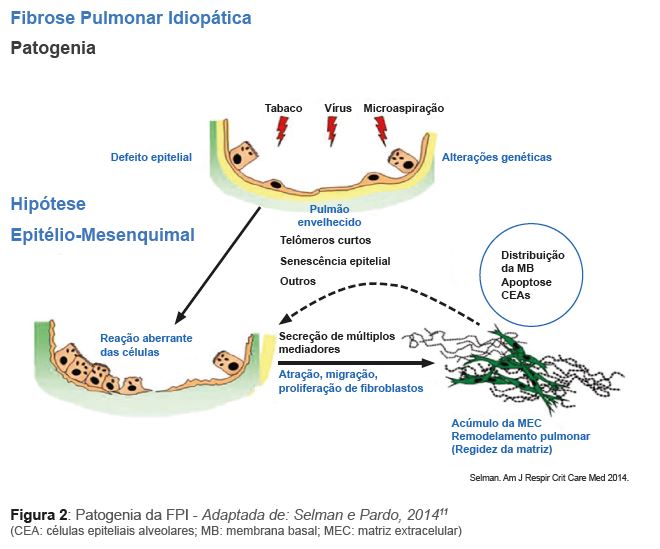
A FPI difere das demais DPIs por resultar de uma agressão ao epitélio alveolar, seguida de um reparo aberrante envolvendo a deposição de colágeno, que se autoperpetua.11 Fatores facilitadores são envolvidos na patogenia da doença, sendo os principais os efeitos decorrentes da senescência,
do tabagismo, do refluxo gastroesofágico (RGE), das mutações e polimorfismos genéticos, além das exposições ambientais, tais como o pó de madeira. Um esquema da patogenia da FPI é mostrado, de maneira simplificada, na figura 2.
O curso clínico da FPI é altamente variável. Há uma população significativa de pacientes que têm um curso mais lento e menos agressivo da doença associado a um tempo de sobrevida mais longo. Em outros o curso da doença pode mudar, com alguns pacientes inicialmente apresentando
doença lenta e estável e, posteriormente, progredindo com rápido declínio da função pulmonar. No curso da doença ainda podem ocorrer exacerbações agudas, caracterizadas por piora rápida da dispneia e maior perda funcional, com surgimento de áreas de vidro fosco ou consolidações na tomografia de tórax de alta resolução (TCAR), que expressam em geral um dano alveolar difuso subjacente de causa indeterminada.12 A mortalidade nestes episódios situa-se em torno de 60%.13 Exacerbação aguda é a causa mais comum de morte na FPI.
Essa grande variabilidade na evolução faz com que o estadiamento e o prognóstico da FPI sejam um desafio. Diversos trabalhos vêm sendo publicados na tentativa de auxiliar o pneumologista na predição prognóstica dos pacientes portadores de FPI. Alguns preditores individuais de sobrevida já estão bem estabelecidos como: maior grau de dispneia na apresentação inicial, hipocratismo digital, presença de hipertensão pulmonar, maior extensão de fibrose na TCAR e número de internações hospitalares, principalmente de causa respiratória.12, 14, 15
A função pulmonar tem papel fundamental na avaliação inicial e para acompanhamento desses pacientes.12, 15 O padrão clássico observado é o distúrbio ventilatório restritivo, com redução da capacidade pulmonar total e da capacidade vital, com relação volume expiratório no primeiro segundo (VEF1)/CVF na faixa prevista ou elevada.
Variáveis de prognóstico:
Quando elevada, a relação VEF1/CVF aponta para um rápido esvaziamento pulmonar, o qual se correlaciona com maior grau de fibrose e pior prognóstico. Valores basais de capacidade vital forçada (CVF) < 65% e da difusão de monóxido de carbono (DCO) < 40% ditam uma pior sobrevida.16-19 A queda longitudinal da CVF em 10% e da DCO em 15%, ao longo do acompanhamento de 3 a 6 meses, também está relacionada a um pior prognóstico.20, 21 Declínio de 5% da CVF medida após 3 meses da avaliação inicial é indicativo de pior prognóstico nesses pacientes.22 DCO é o parâmetro funcional que melhor se correlaciona com a extensão da doença. A saturação periférica de oxigênio medida por oximetria digital (SpO2) < 89% em repouso, ou SpO2 < 85% ao final de teste de exercício, indicam doença avançada.23 Medida da SpO2 pode ser feita em consultório, através de um teste de degrau, em que se solicita ao paciente que desça e suba um degrau em ritmo suficiente para conseguir manter o esforço por quatro minutos. Após três minutos, com a estabilidade do consumo de O2, a SpO2 pode ser obtida.24 Idealmente um teste de caminhada de seis minutos deve ser feito, anotando-se, além da SpO2 ao final do teste, a distância percorrida. Em um estudo realizado no Brasil, distância percorrida abaixo de 330 metros foi indicativa de mau prognóstico.25
Não raramente, devido ao tabagismo, a FPI se associa com enfisema pulmonar, o que potencializa a dispneia pela adição de maior espaço-morto a ser ventilado.26 Um padrão funcional particular é observado nestes casos: a relação VEF1/CVF pode ser normal ou reduzida, porém a CVF é mais
preservada e a troca gasosa é profundamente afetada, expressando-se por maior redução da DCO e maior queda da SpO2 aos esforços. Isto se deve aos efeitos opostos da fibrose e do enfisema sobre a mecânica pulmonar, com efeitos somatórios sobre a piora da troca gasosa.
Outro modo de avaliar o prognóstico inclui o uso de modelos estatísticos compostos de predição ou sistemas de pontuação que tentam combinar variáveis funcionais, dados clínicos e outros exames com valor prognóstico. Esses modelos têm se mostrado mais efi cazes em predizer sobrevida
do que variáveis individuais. Com essa proposta, alguns estudos surgiram nos últimos anos.4, 27-29 Recentemente foi desenvolvido dois estudos com duas coortes brasileiras bem documentadas de FPI na UNIFESP, antes da era antifi brótica.4, 23 No primeiro estudo foi avaliado o prognóstico de
120 pacientes através de: dados clínicos e demográfi cos, medida da dispneia (através de uma das dimensões da escala multidimensional Índice de Dispneia Basal de Mahler, que é a Magnitude da Tarefa) e dados funcionais disponíveis da visita inicial referente aos exames de espirometria, DCO
e SpO2 de repouso e exercício.4 E no segundo estudo, com o mesmo desenho, 173 casos que não possuíam DCO foram avaliados.23 Os resultados são semelhantes com e sem medida da DCO, disponível apenas em centros maiores, e ajudam muito na avaliação inicial dos pacientes (tabela 1).

REFERÊNCIAS:
1. Baldi BG, Pereira CA, Rubin AS, Santana AN, Costa AN, Carvalho CR, et al. Highlights of
the Brazilian Thoracic Association guidelines for interstitial lung diseases. J Bras Pneumol.
2012;38(3):282-91.
2. Baddini-Martinez J, Ferreira J, Tanni S, Alves LR, Cabral Junior BF, Carvalho CRR, et al.
Brazilian guidelines for the pharmacological treatment of idiopathic pulmonary fibrosis. Official
document of the Brazilian Thoracic Association based on the GRADE methodology. J Bras
Pneumol. 2020;46(2):e20190423.
3. Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of
Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J
Respir Crit Care Med. 2018;198(5):e44-e68.
4. Soares MR, Pereira C, Ferreira R, Nei Aparecida Martins Coletta E, Silva Lima M, Muller Storrer
K. A score for estimating survival in idiopathic pulmonary fibrosis with rest SpO2>88. Sarcoidosis
Vasc Diffuse Lung Dis. 2015;32(2):121-8.
5. Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/
ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and
management. Am J Respir Crit Care Med. 2011;183(6):788-824.
6. Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic
pulmonary fibrosis: a systematic review. Eur Respir J. 2015;46(3):795-806.
7. Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary
fibrosis: review of the literature. Eur Respir Rev. 2012;21(126):355-61.
8. Baddini-Martinez J, Pereira CA. How many patients with idiopathic pulmonary fibrosis are there
in Brazil? J Bras Pneumol. 2015;41(6):560-1.
9. Thannickal VJ, Murthy M, Balch WE, Chandel NS, Meiners S, Eickelberg O, et al. Blue
journal conference. Aging and susceptibility to lung disease. Am J Respir Crit Care Med.
2015;191(3):261-9.
10. Spagnolo P, Rossi G, Cavazza A. Pathogenesis of idiopathic pulmonary fibrosis and its clinical
implications. Expert Rev Clin Immunol. 2014;10(8):1005-17.
11. Selman M, Pardo A. Revealing the pathogenic and aging-related mechanisms of the enigmatic
idiopathic pulmonary fibrosis. an integral model. Am J Respir Crit Care Med. 2014;189(10):1161-72.
12. Ley B, Collard HR, King TE. Clinical course and prediction of survival in idiopathic pulmonary
fibrosis. Am J Respir Crit Care Med. 2011;183(4):431-40.
13. Collard HR, Ryerson CJ, Corte TJ, Jenkins G, Kondoh Y, Lederer DJ, et al. Acute Exacerbation
of Idiopathic Pulmonary Fibrosis. An International Working Group Report. Am J Respir Crit Care
Med. 2016;194(3):265-75.
14. Brown AW, Fischer CP, Shlobin OA, Buhr RG, Ahmad S, Weir NA, et al. Outcomes after
hospitalization in idiopathic pulmonary fibrosis: a cohort study. Chest. 2015;147(1):173-9.
15. Nathan SD, Meyer KC. IPF clinical trial design and endpoints. Curr Opin Pulm Med. 2014.
16. du Bois RM, Weycker D, Albera C, Bradford WZ, Costabel U, Kartashov A, et al. Forced vital
capacity in patients with idiopathic pulmonary fibrosis: test properties and minimal clinically
important difference. Am J Respir Crit Care Med. 2011;184(12):1382-9.
17. Lama VN, Flaherty KR, Toews GB, Colby TV, Travis WD, Long Q, et al. Prognostic value of
desaturation during a 6-minute walk test in idiopathic interstitial pneumonia. Am J Respir Crit
Care Med. 2003;168(9):1084-90.
18. Stephan S, de Castro Pereira CA, Coletta EM, Ferreira RG, Otta JS, Nery LE. Oxygen
desaturation during a 4-minute step test: predicting survival in idiopathic pulmonary fibrosis.
Sarcoidosis Vasc Diffuse Lung Dis. 2007;24(1):70-6.
19. Mogulkoc N, Brutsche MH, Bishop PW, Greaves SM, Horrocks AW, Egan JJ, et al. Pulmonary
function in idiopathic pulmonary fibrosis and referral for lung transplantation. Am J Respir Crit
Care Med. 2001;164(1):103-8.
20. Collard HR, King TE, Bartelson BB, Vourlekis JS, Schwarz MI, Brown KK. Changes in clinical
and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care
Med. 2003;168(5):538-42.
21. Flaherty KR, Thwaite EL, Kazerooni EA, Gross BH, Toews GB, Colby TV, et al. Radiological
versus histological diagnosis in UIP and NSIP: survival implications. Thorax. 2003;58(2):143-8.
22. Russell AM, Adamali H, Molyneaux PL, Lukey PT, Marshall RP, Renzoni EA, et al. Daily Home
Spirometry: An Effective Tool for Detecting Progression in Idiopathic Pulmonary Fibrosis. Am J
Respir Crit Care Med. 2016;194(8):989-97.
23. Fukuda CY, Soares MR, Pereira CAC. A Score Without DLCO for Estimating Survival in Idiopathic
Pulmonary Fibrosis. Am J Resp Crit Care Med. 2018;197:A2535.
24. Dal Corso S, Duarte SR, Neder JA, Malaguti C, de Fuccio MB, de Castro Pereira CA, et al. A
step test to assess exercise-related oxygen desaturation in interstitial lung disease. Eur Respir
J. 2007;29(2):330-6.
25. Mancuzo EV, Soares MR, Pereira CAC. Six-minute walk distance and survival time in patients
with idiopathic pulmonary fibrosis in Brazil. J Bras Pneumol. 2018;44(4):267-72.
26. Costa CM, Neder JA, Verrastro CG, Paula-Ribeiro M, Ramos R, Ferreira EM, et al. Uncovering
the mechanisms of exertional dyspnoea in combined pulmonary fibrosis and emphysema. Eur
Respir J. 2020;55(1).
27. Ley B, Ryerson CJ, Vittinghoff E, Ryu JH, Tomassetti S, Lee JS, et al. A multidimensional index
and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156(10):684-91.
28. Wells AU, Desai SR, Rubens MB, Goh NS, Cramer D, Nicholson AG, et al. Idiopathic pulmonary
fibrosis: a composite physiologic index derived from disease extent observed by computed
tomography. Am J Respir Crit Care Med. 2003;167(7):962-9.
29. du Bois RM, Weycker D, Albera C, Bradford WZ, Costabel U, Kartashov A, et al. Ascertainment
of individual risk of mortality for patients with idiopathic pulmonary fibrosis. Am J Respir Crit
Care Med. 2011;184(4):459-66.
30. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of
nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071-82.
Sorry, the comment form is closed at this time.