CRISES EPILÉPTICAS E EPILEPSIAS: esquema diagnóstico
Profa. Dra. Elza Márcia Yacubian CRM-SP 27.653
Professora Adjunta do Depar-tamento de Neurologia e Neu-rocirurgia da Universidade Federal de São Paulo (Unifesp).
O esquema diagnóstico
O esquema diagnóstico para a Classificação das Epilepsias oferece a possibilidade de diagnóstico em múltiplos níveis, dependendo da informação e dos recursos disponíveis11 (Figura2). Nele, o primeiro passo (nível 1) consiste em estabelecer se um determinado evento paroxístico é uma crise epiléptica. Uma vez que esse diagnóstico tenha sido estabelecido clinicamente (ou por meio de exames auxiliares, como EEG, vídeo-EEG ou ambos), o próximo pas-so será classificar o(s) tipo(s) de crise(s). Algumas vezes o diagnóstico precisará ser interrompido nesse nível, pois em algumas situações, como quando estamos diante de uma primeira crise epiléptica, não será possível prosseguir para os próximos níveis.
Na maioria das vezes, no entanto, será possível chegar ao nível 2, ou seja, tentar classificar a epilepsia com base no(s) tipo(s) de crise(s). No nível 2, as epilepsias deverão ser classificadas como focais, generalizadas, focais e generalizadas (quando ambos os tipos de crises estiverem presentes) ou desconhecidas (quando for impossível
classificar as crises como focais ou generalizadas).
No próximo passo (nível 3), vamos tentar estabelecer o diagnóstico de uma síndrome epiléptica. Uma síndrome epiléptica é um conjunto de características clínicas, eletroencefalográficas, imagenológicas e etiológicas. Esse diagnóstico terá muita importância para o tratamento e o estabelecimento do prognóstico.
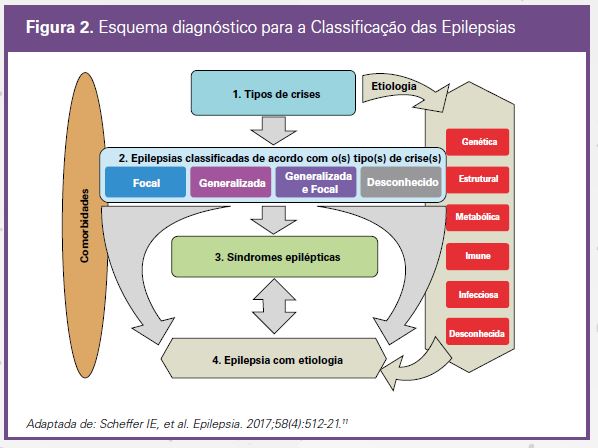
Embora o Esquema Diagnóstico enfatize em todos os seus três níveis que é fundamental estabelecer a etiologia da epilepsia (no Esquema Diagnóstico à direita, na barra vertical, estão os seis grupos etiológicos), é o quarto nível (Nível 4) que define o diagnóstico da epilepsia e sua etiologia. Em algumas circunstâncias, mesmo sem o estabele-cimento da síndrome epiléptica, é possível estabelecer o diagnóstico etiológico. Um exemplo de uma dessas condições é a definição de que a etiologia da epilepsia de um determinado paciente é uma mutação na subunidade alfa-1 do canal de sódio (SCN1A), a qual é encontrada em um espectro de manifestações clínicas de gravidade crescente, desde crises febris simples até a síndrome de Dravet, no extremo mais grave desse espectro.
Embora em todos os níveis nossa atenção deva estar voltada para o estabelecimento da etiologia da epilepsia, infe-lizmente, em vários deles, a etiologia não poderá ser estabelecida a despeito de todos os nossos esforços. Em outros casos, verificaremos mais de uma etiologia para uma mesma epilepsia. Assim, por exemplo, a epilepsia pode ter duas etiologias, por exemplo, uma estrutural e outra genética, como é o caso da esclerose tuberosa. Nesta, ambas as etiologias acarretam implicações terapêuticas fundamentais, como a ressecção da lesão estrutu-ral, ou seja, de um túber, ou o uso de inibidores da mTOR (mammalian target of rapamycin – alvo da rapamicina em mamíferos) no tratamento medicamentoso que promoverá uma interferência na via do distúrbio genético.
Finalmente, encerrando o Esquema Diagnóstico, pacientes com epilepsia podem apresentar uma gama de comorbi-dades ampla (representadas na elipse à esquerda), as quais podem ser encontradas em qualquer forma das doen-ças epilépticas e também podem contribuir para o diagnóstico etiológico. Assim, por exemplo, meninas com muta-ções no gene PCDH-19, que produz a proteína protocaderina-19 (PCDH-19), apresentam alterações comportamen-tais com características do espectro autista e episódios de terror, os quais são, de longe, mais graves do que as crises epilépticas per se.
Para a melhor compreensão da terminologia utilizada nesse Esquema Diagnóstico, foram ainda definidos alguns termos importantes nele utilizados ou frequentemente empregados na caracterização das epilepsias.
Genético
Até agora, uma mutação genética é reconhecida em algumas poucas formas de epilepsiase uma mesma mutação pode determinar várias síndromes epilépticas. Em poucas dessas síndromes as mutações são familiares. A maioria delas é de mutações de novo, ou seja, que ocorrem apenas em um indivíduo de uma família. Assim, o termo genético não é sinônimo de hereditário e, na grande maioria das epilepsias, não há ainda demonstração inequívoca do fator genético determinante da doença.11
Na nova classificação, o termo idiopático empregado na Classificação de 1989 não foi utilizado. Como se referir a esse grupo de epilepsias?
O termo idiopático foi usado na classificação das etiologias das epilepsias de 1989, quando a etiologia de uma forma de epilepsia era “presumivelmente genética”. Nele se enquadravam as epilepsias generalizadas idiopáticas, um grupo muito importante, pois contribui para 25% de todas as epilepsias e é constituído pela epilepsia ausência da infância, epilepsia ausência juvenil, epilepsia mioclônica juvenil e epilepsia apenas com crises tônico-clônicas generalizadas. Nelas, estudos de gêmeos e de famílias com grande número de indivíduos afetados sugerem fortemente um componente genético, embora na maioria dessas síndromes ainda não tenham sido individualiza-dos genes específicos. Para esse grupo, o termo etiologia de origem desconhecida poderia ser utilizado. No entanto, em algumas famílias com essas epilepsias, como na epilepsia mioclônica juvenil, já foram identificados os genes EFHC1 e GABRA1, fazendo, assim, que a epilepsia dessas famílias seja mais bem definida como de etiologia genética. Ainda se busca uma melhor denominação para esse grupo de síndromes epilépticas, por exemplo, epilep-sias generalizadas genéticas ou epilepsias generalizadas de etiologia genética ou desconhecida.11
Encefalopatia epiléptica e/ou do desenvolvimento
Esse termo tem sido amplamente utilizado na área de epilepsia nas últimas décadas. A definição de Berg et al., em 2010,10 introduziu a noção de que, em algumas epilepsias, a atividade epileptiforme per se contribui para compro-metimentos cognitivos e comportamentais acima e além do que os que seriam esperados pela patologia subja-cente. Esses comprometimentos, sejam eles seletivos ou globais, se expressariam em um espectro de gravidade e poderiam piorar com o passar do tempo.
Desde a década de 1970 se reconhece, especialmente em lactentes e crianças pequenas, que, enquanto a presen-ça de atividade epileptiforme abundante promove alentecimento cognitivo e regressão no desenvolvimento associ-ados a consequências psiquiátricas e comportamentais, a melhora desse padrão eletroencefalográfico pode promover melhor prognóstico, por prevenir as consequências deletérias observadas nesse grupo de epilepsias em um estágio crítico do desenvolvimento neurológico. Há algumas síndromes em que esse fato definitivamente ocor-re, como nas clássicas síndromes de West e de Lennox-Gastaut, mas há algumas encefalopatias mais recentemen-te reconhecidas nas quais também se evidencia claramente essa associação, como nas encefalopatias CDKL5 (síndrome de Rett atípica) e CHD2. Em outras, contudo, muito provavelmente, apenas a presença de atividade epileptiforme não é suficiente para deflagrar vários desses prejuízos. Assim, na síndrome de Dravet, uma epilepsia tida como um protótipo das “encefalopatias epilépticas”, a regressão neurológica e comportamental já é evidente entre 1 e 2 anos de idade, em uma época em que as alterações eletroencefalográficas ainda não estavam presen-tes ou eram pouco importantes. Nessa doença há, portanto, dois componentes: o primeiro relativo ao desenvolvi-mento e o segundo, epiléptico, ambos determinados, em 80% dos casos, por alguma das mais de mil mutações
hoje conhecidas no gene SCN1A. Ao descrever esse grupo de doenças epilépticas, podem-se utilizar os termos, iso-ladamente, encefalopatia epiléptica e encefalopatia do desenvolvimento ou, conjuntamente, encefalopatia epilép-tica e/ou do desenvolvimento.11
Como se referir ao termo epilepsias generalizadas sintomáticas da classificação de 1989?
Essas formas de epilepsia, como a síndrome de Lennox-Gastaut, se enquadram hoje no grupo das encefalopatias epilépticas e/ou do desenvolvimento, indicando que o termo epilepsias generalizadas sintomáticas deve ser aban-donado.11 Contudo, aquelas raras epilepsias com retardo no desenvolvimento estável e crises epilépticas refratá-rias poderiam ser descritas como atraso intelectual com epilepsia focal, generalizada ou ainda focal e generaliza-da.
Epilepsias benignas
A proposta de Organização das Crises Epilépticas e Epilepsias de 2010 já sugeria o abandono do termo benigno.10 De fato, nessas epilepsias há diferentes graus de comorbidades, desde dificuldades de aprendizado a transtornos do espectro autista. O termo benigno subestimaria a gravidade dessas comorbidades. Foram sugeridas as denomi-nações epilepsias autolimitadas, indicando que elas se resolvem com o tempo, ou epilepsias fármaco-responsivas, pois nelas as crises são facilmente controladas com o uso de fármacos antiepilépticos (FAEs) adequados.10,11
A classificação das crises epilépticas como guia para o tratamento
O último passo é o estabelecimento da terapêutica. A figura 3 mostra o espectro de ação dos FAEs disponíveis no Brasil para o tratamento das crises epilépticas.
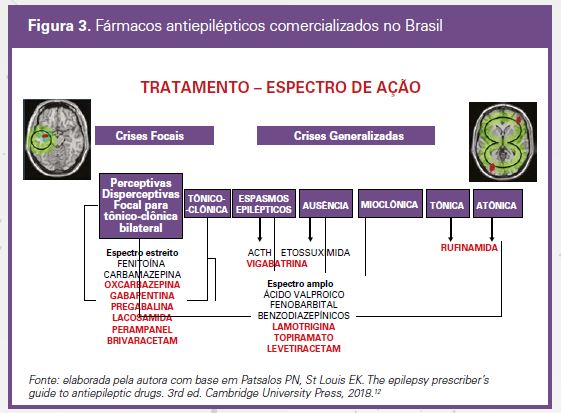
O arsenal para tratamento das crises epilépticas e das epilepsias, inicialmente constituído por poucos FAEs tradi-cionais, foi acrescido, após a década de 1990, de mais de uma dezena de novos medicamentos aprovados pelas autoridades regulatórias para diferentes tipos de crises e perfis de pacientes. Nos últimos anos, foram introduzidos
no Brasil nove novos FAEs: oxcarbazepina, lamotrigina, topiramato, gabapentina, pregabalina, vigabatrina, lacosami-da, levetiracetam e brivaracetam. Por seu melhor perfil farmacocinético, vários dos novos FAEs têm encontrado in-dicação visando à prevenção das consequências deletérias a curto e longo prazo de um tratamento crônico, como o das epilepsias, e também em pacientes com comorbidades, como alterações hepáticas e renais, obesidade, cefa-leia e transtornos do humor. Em todas essas situações, sua indicação deveria ser considerada em monoterapia e, algumas vezes, como primeira opção terapêutica.
Confira a primeira parte desse artigo clicando aqui.
Referências
1. Thurman DJ, Beghi E, Begley CE, Berg AT, Buchhalter JR, Ding D, et al.; ILAE Commission on Epidemiology. Standards for epidemiologic studies and surveillance
of epilepsy. Epilepsia. 2011;52 Suppl 7:2-26. 2. Hauser WA, Annegers JF, Kurland LT. Prevalence of epilepsy in Rochester, Minnesota: 1940-1980. Epilepsia.
1991;32(4):429-45. 3. Fisher RS, van Emde Boas W, Blume W, Elger C, Genton P, Lee P, et al. Epileptic seizures and epilepsy: definitions proposed by the International
League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia. 2005;46(4):470-2. 4. Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A,
Cross JH, Elger CE, et al. A practical clinical definition of epilepsy. Epilepsia. 2014;55(4):475-82. 5. Blume WT, Lüders HO, Mizrahi E, Tassinari C, van Emde Boas W,
Engel J Jr. Glossary of Descriptive Terminology for Ictal Semiology: Report of the ILAE Task Force on Classification and Terminology. Epilepsia. 2001;42(9):1212-8.
6. Hauser AW, Rich SS, Lee JR, Annegers JF, Anderson VE. Risk of recurrent seizures after two unprovoked seizures. N Engl J Med. 1998;338(7):429-34. 7. Proposal
for revised clinical and electroencephalographic classification of epileptic seizures. From the Commission on Classification and Terminology of the International League
Against Epilepsy. Epilepsia. 1981;22(4):489-501. 8. Fisher R, Cross H, French JA, Higurashi N, Hirsch E, Jansen FE, et al. Operational classification of seizure types
by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58(4):522-30. 9. Proposal for
revised classification of epilepsies and epileptic syndromes. Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia.
1989;30(4):389-99. 10. Berg AT, Berkovic SF, Brodie M, Buchhalter J, Cross JH, van Emde Boas W, et al. Revised terminology and concepts for organization of
seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia. 2010;51(4):676-85. 11. Scheffer IE, Berkovic S,
Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology.
Epilepsia. 2017;58(4):512-21. 12. Patsalos PN, St Louis EK. The epilepsy prescriber’s guide to antiepileptic drugs. 3rd ed. Cambridge University Press, 2018.
Sorry, the comment form is closed at this time.